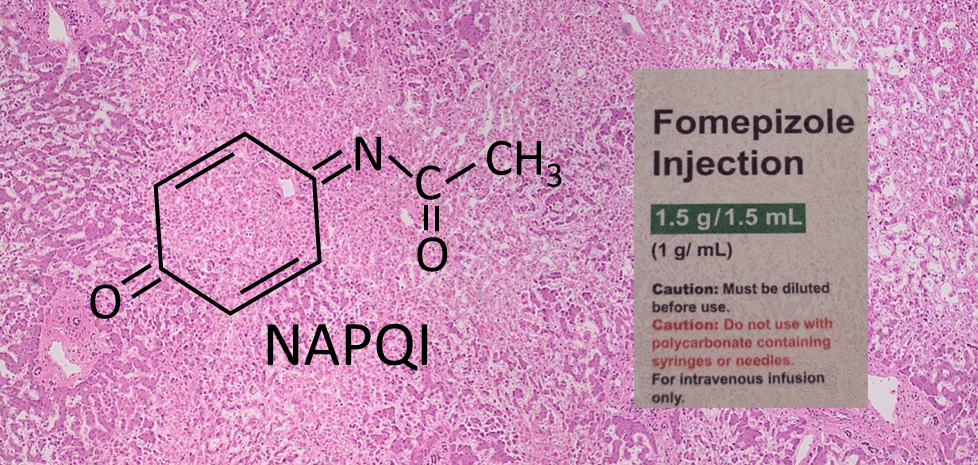
Tox and Hound – Fellow Friday
What Terror Hath NAPQI Wrought?
From NAPQI to Hepatic Necrosis to Fomepizole
Steven Curry, M.D.
University of Arizona College of Medicine – Phoenix
Department of Medical Toxicology
Banner – University Medical Center Phoenix
Phoenix, AZ
Introduction
Medical toxicology fellows can recite the routes of acetaminophen (APAP) metabolism in their sleep. And they know that after overdose, the total amount of APAP undergoing oxidative metabolism by CYP2E1 to form NAPQI depletes glutathione (GSH). NAPQI then covalently binds to proteins to begin a series of bad things that culminate in cell necrosis (Figure 1).

This post is about the bad things subsequent to NAPQI formation, those events that fellows really don’t know or read much about in standard texts and resources. An enormous amount of research using animals, cell cultures, and in vitro assays, along with data from human studies abound in the literature. Importantly, results from such studies offer opportunities beyond N-acetylcysteine (NAC) to prevent liver injury and failure and explain the excitement about possible reversal of JNK activation in the late treatment of APAP toxicity in humans.
Rob Hendrickson has appropriately condensed a cascade of events into a couple paragraphs in his comprehensive chapter on APAP toxicity in Goldfrank’s. I might have made it even shorter for a text. This post, however, will greatly expand on that cascade with attempts to simplify and explain concepts for purposes of understanding. Yet even this discussion must be very abbreviated and simplified.
A special thanks to Hartmut Jaeschke and Anup Ramachandran from University of Kansas who have been generous in sharing several illustrations that I have significantly modified, but which saved weeks of time in preparation of this post. Hartmut has always been the expert to whom I first turn regarding cellular events of APAP toxicity, and has been answering my questions for decades, quite literally. Mitch McGill, formerly at University of Kansas, but now at University of Arkansas, has also responded to ASK1 and JNK inquires and dinner invitations over the years.
By the end of this post, medical toxicology fellows will recognize and understand:
- Similar APAP toxicity chain of events in mouse models and human beings
- Initial NAPQI-induced toxicity characterized by superoxide release from electron transport chain (ETC) complex III
- Activated JNK produces superoxide release from ETC complex I
- An amplified oxidative/nitrosative cycle maintains persistent JNK activation and peroxynitrite formation
- Peroxynitrite’s important role in producing hepatocellular necrosis
- Fomepizole demonstrates 2 mechanisms of action: inhibition of CYP2E1, and inhibition of JNK
- Fomepizole’s inhibition of JNK prevents necrosis in mice and human liver cells long after NAPQI formation and GSH depletion
It looks complicated, but it is easy to understand if we start at the beginning and build on each step. Let’s begin.
Models
If only human studies could be used to establish the chain of events leading to hepatocyte necrosis in man following APAP OD, we would require that liver biopsies be performed as often as every 30 to 60 minutes for several days in groups of subjects with identical, single, known ingested APAP doses and known ingestion times. Some groups would need to be treated with various chemicals that would affect intracellular pathways pertaining to necrosis. Perhaps we could find groups of patients with identical genetic alterations in various detoxification pathways to clarify the importance of those systems. Hundreds of experiments involving tens of thousands of subjects or more would be required. Good luck, even if you get funding.

Thus, investigators use small animal models that, along with studies in human cell cultures and overdose patients, are thought to most closely represent what occurs in human beings. The mouse model has been the most commonly used. In fact, Mitchell used mice in his four papers that first set forth P450-generated NAPQI formation, GSH depletion, and covalent binding. The mouse model also first demonstrated NAC as an antidote for APAP toxicity. We also know that rats are poor models because of resistance to toxicity and different pathways leading to necrosis.
But mice are not human beings. For example, different strains (and sex) of mice vary in their response to APAP OD. The rapid absorption of an intraperitoneal dose of APAP in a mouse contrasts with gradual absorption, sometimes extending over more than a day, in human overdoses. Most important, the entire time course of events in most mouse studies is dramatically compressed compared to humans. Absorption of an intraperitoneal APAP dose, metabolism, all NAPQI formation, and maximal GSH depletion commonly occur within 30 minutes. Peak ALT elevation may occur within 6 hours, depending on the dose. Resolution of necrosis in surviving mice occurs over 24 to 96 hours. While mouse experiments provide data suggesting a sequence of events leading to necrosis (and recovery) in man, we cannot immediately extrapolate the time course of such events to humans.

Cell culture studies involving human hepatocytes provide helpful information, but present their own limitations. As examples, gene expression in cultured hepatocytes changes over time. CYP2E1 expression decreases after a couple days in mouse and human hepatocyte cultures, so studies must be performed on freshly cultured cells. Expression of genes controlling antioxidant pathways change over time. Most cell cultures are performed using room air (pO2 ~ 158 mm Hg), while the pO2 experienced by centrilobular hepatocytes in vivo is about 30 mm Hg. Fortunately, studies have shown that while the rate of APAP-induced necrosis in cell cultures is accelerated at room air compared with lower oxygen tensions, steps leading to necrosis appear to be similar to in vivo studies. Man-made liver organoids also fail to experience APAP-induced necrosis to date because of loss of most CYP2E1 expression as the cultures grow over the extended time required for organoid formation.
This post first will summarize the events leading to hepatocyte necrosis based on animal experiments, hepatocyte cultures, and in vitro studies involving mice. Then it will examine the extent to which results from mouse studies support similar events in human beings. Finally, with an understanding of cellular events subsequent to NAPQI, the rationale for potential therapies like fomepizole that may prevent or attenuate hepatocyte death long after NAPQI formation will be addressed.
Figure 4 generally illustrates where we are headed and immediately makes apparent that NAPQI-induced hepatocyte necrosis centers on mitochondrial events. Thus, we begin with a brief foundation of mitochondrial structure, electron transport, oxidative phosphorylation, and generation of reactive oxygen species that may have slipped the memory of some readers. This foundation builds on previous discussions of mitochondrial function from Fellow Friday posts on cyanomythology, the pathogenesis of microvesicular steatosis, lactic acidosis, and phosphine toxicity, in which oxidative phosphorylation and other biochemical reactions within the matrix were discussed.

Mitochondria
Each hepatocyte contains 500 to 2,000 mitochondria, and each mitochondrion contains an average of 5 circular mDNA molecules. Figure 5 shows a photomicrograph of a single hepatocyte treated with a marker that causes each of countless mitochondria to fluoresce as white dots.

Mitochondria contain two lipid bilayer membranes (Figure 6). The outer membrane is traversed with porins (voltage-dependent anion channel [VDAC]) and is permeable to small molecules. No voltage gradient across the outer membrane with respect to cytosol has been demonstrated. Nuclear-coded proteins have specific tags that allow them to be imported across the outer membrane.

The inner mitochondrial membrane is highly selective as to what will cross and is embedded with ion channels, SLC proteins, and other transporters responsible for translocating various molecules. Small lipophilic molecules can still diffuse across. Invaginations of the inner membrane into the matrix called cristae are embedded with components of the electron transport chain (ETC) and ATP synthase. The intermembrane space resides between the inner and outer membranes.
The inner membrane encompasses the matrix, containing enzymes and cofactors needed for a multitude of reactions, including the tricarboxylic acid cycle, the urea cycle, fatty acid oxidation, heme synthesis and other pathways. The protein content of the matrix is so high that its consistency is similar to paste. Most mitochondrial proteins are encoded by nuclear DNA, and specific N-terminal tags allow importation of proteins across the inner membrane, as well. Mitochondrial DNA codes for a portion of proteins used in electron transport complexes I, III and IV and for tRNAs and rRNAs.
Electron transport and oxidative phosphorylation
Take a look at the illustration of the inner membrane in Figure 7.

NADH generated in the Krebs cycle and other sources increases the NADH/NAD+ ratio. Thus, NADH donates two electrons onto complex I in order to move the NADH/NAD+ mass action ratio back towards equilibrium. Electrons then move from complex I onto Q (ubiquinone). Similarly, electrons from succinate or FADH2 (through an intermediate) arising from fatty acid oxidation move from complex II onto Q. From Q, electrons are shuttled to complex III, then to cytochrome c (residing on the external surface of the inner membrane), and finally onto complex IV (cytochrome oxidase), where they combine with O2 to create H2O. As electrons move down the ETC and eventually onto O2, released energy is used to create a proton gradient across the inner membrane, with a lower pH (higher proton concentration) and more positive charge (voltage gradient) in the intermembrane space than matrix. A typical voltage potential across the inner membrane is about -180 mV (matrix negative). At steady state, the amount of energy found in the increased NADH/NAD+ ratio is about equal to the energy stored across the inner membrane as voltage and proton concentration (pH) gradients. Those protons re-enter the matrix by traveling through a pore in ATP synthase, and the released energy is used to make ATP, increasing the ATP/ADP ratio. Among structures not illustrated in Figure 7 are the adenine nucleotide transporter (ANT), which exchanges ATP in the matrix for ADP in the intermembrane space, and an importer for phosphate.
Generation of reactive oxygen species (ROS)
Important steps in the pathogenesis of NAPQI-induced hepatic necrosis involve creation of superoxide (O2.-) and hydrogen peroxide (H2O2), as well as peroxynitrite. For now, we will simply observe that if electron transport is inhibited for some reason, yet the NADH/NAD+ ratio remains significantly elevated above it’s equilibrium mass action ratio, then electrons may leave the electron transport chain and move onto O2 to create O2.- Other physiologic changes in mitochondria may also increase ROS generation without necessarily dramatically affecting oxidative phosphorylation. Electrons most commonly depart complex I, Q, and complex III. Complex I only directs superoxide toward the matrix, while complex III can direct O2.- toward the intermembrane space or the matrix (as demonstrated by using various agents that inhibit electron transport at different locations within complex III). Mn superoxide dismutase (MnSOD) in the matrix and Cu/ZnSOD in the intermembrane space and cytosol convert O2.- to H2O2.
While abnormally elevated concentrations of H2O2 can be profoundly toxic, H2O2 is an important signaling molecule generated by the ETC and a plethora of enzymatic reactions. It is normally present at low concentrations within cells, and it is estimated that 0.1% to 0.2% of hepatic oxygen consumption normally represents electron leak from the ETC to form ROS.
NAPQI and Complex III: First ROS Release
We now begin with NAPQI and examine subsequent steps to hepatocyte necrosis in mice. NAPQI is a short-lived and soft electrophile with an estimated intracellular half-life of < 7 seconds before combining with GSH or cysteine and lysine residues on various proteins. A soft electrophile is one in which the positive charge is distributed throughout the molecule, allowing it to remain lipophilic and diffuse across lipid membranes.
CYP2E1 historically was believed to reside only bound to membranes of smooth endoplasmic reticulum (SER). One could imagine that some finite fraction of NAPQI would survive long enough to diffuse into mitochondria. In fact, circulating adducts between hemoglobin and NAPQI have been detected, indicating some NAPQI even escapes hepatocytes, diffuses into RBCs, and combines with hemoglobin during its short existence.
However, it is now recognized that CYP2E1 also resides in mitochondria, bound to the inner membrane, where it forms NAPQI from APAP. An estimated 10 to 40% of microsomal (membrane-associated) CYP2E1 represents mitochondrial CYP2E1 in mice. Of course, CYP2E1 is mainly expressed in hepatocytes in the several cell layers around the central vein (Figure 8). Centrilobular hepatocytes also have lower GSH concentrations than those in the midzonal and periportal areas. These, and other differences in gene expression between zones explain the propensity for centrilobular necrosis following APAP OD.

While NAPQI binds to many different hepatocyte proteins, various experiments have demonstrated that binding to a mitochondrial target(s) is essential to begin the chain of events leading to necrosis. In Figure 9, NAPQI formed in SER and/or mitochondria binds to a protein(s) in or associated with complex III of the ETC to allow electrons to combine with O2 to create O2.-, which is directed into the intermembrane space. Cu/ZnSOD converts some superoxide to H2O2, which diffuses into the cytosol. Unreacted O2.- moves through voltage-dependent anion channels (VDAC) into the cytosol, where some is also enzymatically converted to H2O2.

The extent to which superoxide generated by complex III in response to NAPQI is a consequence of some degree of inhibition of electron transport is not completely understood, but possible. NAPQI binding and subsequent superoxide generation are initially associated with a modest drop in voltage potential across the inner membrane without an increase in oxygen consumption or proton leak – i.e., without uncoupling of oxidative phosphorylation.
JNK Activation and Translocation to Mitochondria
First is a brief interlude reminder on the subject of mitogen-activated protein kinases (MAPKs), which undoubtedly was everyone’s favorite topic in medical school. MAPKs are a group of enzymes which phosphorylate serine or threonine residues. These enzymes act through cascades in which one enzyme activates a different kinase via phosphorylation, and so forth. Cascades allow for signal amplification and various locations for other pathways or signals to enter and up- or down-regulate cascade activity.
MAPK pathways are typically divided into groups of 3 MAPKs (Figure 10). The MAP3K member senses a signal (e.g. oxidant stress) to become activated and begins the cascade. The final member, termed an MAPK, phosphorylates a target resulting in some effect. In our simplified overview, we are interested in the MAP3K members named apoptosis signal-regulating kinase 1 and 2 (referred to singly as ASK1/2), the MAP2K member named MKK4, and the MAPK member named c-Jun N-terminal kinase (JNK). Of the 3 subtypes of JNK, JNK1 and JNK2 are most relevant to our interest, and they will be collectively referred to as JNK. In the absence of cascade activation, MAPK phosphatases keep MKK4 and JNK inactive through dephosphorylation.

ASK1/2 resides in cytosol, where it is activated by H2O2 released from mitochondria. How would ASK1/2 recognize a rise in H2O2 concentration? While there are many mechanisms by which oxidant stress can be sensed, in this instance it is explained by a thioredoxin.
A thioredoxin is an oxidoreductase protein that binds and inactivates ASK1/2 (or other proteins) through covalent binding involving cysteine residues. When oxidant stress increases, cysteine residues on thioredoxin undergo oxidation and cross-bridging, releasing ASK1/2 from inhibition (Figure 11). ASK1/2 then phosphorylates MKK4, which, in turn, phosphorylates JNK to form active pJNK (phosphorylated JNK). pJNK then translocates to mitochondria and, using ATP, phosphorylates Sab, a scaffold protein confined to, but spanning, the outer membrane.

Second ROS Release and Peroxynitrite Formation
Figure 12 illustrates consequences of Sab phosphorylation. I hope you aren’t tired of phosphorylation because we’ve just begun. A protein kinase named Src resides in the intermembrane space where it is complexed to another protein, DOK4 (not shown), that is bound to the inner membrane. Src normally is phosphorylated and active, which is required for normal functioning of the ETC.
Scaffold proteins bind other proteins, and SHP1, a phosphatase, is bound by Sab in the intermembrane space. Upon Sab’s phosphorylation by pJNK on the outside of the outer membrane, SHP1 is released into the intermembrane space where it undergoes activation through phosphorylation by a kinase, perhaps by pSrc. Activated pSHP1 (a phosphatase) then dephosphorylates and inactivates pSrc, inhibiting the ETC.

The exact site(s) of inhibition of the ETC in response to dephosphorylation of Src has not been proved, though there is evidence to suggest that it may be at the levels of complexes I and II, as these seem to be normal targets for Src kinase activity. Regardless, the eventual consequence of ETC inhibition is an outburst of superoxide from complex I into the matrix (Figure 13).

For our purposes, superoxide in the matrix has two main fates (Figures 13 and 14). A portion of superoxide combines with nitric oxide (NO) to form peroxynitrite (ONOO–). Peroxynitrite is a very strong, short-lived oxidant which nitrates targets, including proteins, membranes, and DNA (Figure 15). The nitration of tyrosine in proteins to form nitrotyrosine is enhanced by ferrous iron. Fe2+ is released from lysosomes into the cytosol before transport into the matrix across MCFU, an electrogenic Ca2+/Fe2+ uniporter (Figure 13). The exact cause of lysosomal iron release in APAP toxicity is not known, but such release is a common event in stressed cells.


Superoxide’s second fate is conversion to H2O2 by MnSOD (Figure 14). H2O2 can react with Fe2+ to form hydroxyl radicals, also very damaging oxidants. But lipid peroxidation of mitochondrial or cell membranes by OH.- does not appear to play an important role in cell necrosis from APAP. More important, H2O2 diffuses into the cytosol where it continues to activate ASK1/2 and, thus, JNK, which causes ROS release from complex I, and the cycle begins again, augmented by ROS from complex III, especially if NAPQI is still being formed (Figure 16 & 17). This vicious positive feedback loop is what is labeled as amplified oxidative/nitrosative stress in Figure 13. This cycle has also been referred to as the pJNK/SAB/ROS activation loop.

This amplified cycle leads to persistent JNK activation and peroxynitrite formation in the face of depleted or inactivated antioxidant protection mechanisms, such as GSH, which normally reduces H2O2, O2.- and ONOO–. GSH levels originally fell from NAPQI, and have yet to be restored. GSH recovery is limited, in part, by reacting with continually produced ROS and peroxynitrite. MAPK phosphatases can dephosphorylate and inactivate kinases in the cascade, but ASK1/2 keeps getting reactivated from the continued oxidant stress, leading to activation of JNK.
But we still have not arrived at necrosis. In fact, a hepatocyte may yet survive if GSH synthesis can catch up and restore GSH levels, and/or if various MAPK phosphatases are finally able to persistently keep enzymes inactivated. Partially damaged mitochondria can even be repaired/removed (e.g., mitophagy). But, what are the consequences of this amplified oxidative stress and peroxynitrite production if it continues?
mDNA
Peroxynitrite dramatically damages and depletes mitochondrial DNA (mDNA). Of course, this impairs synthesis of proteins needed for complexes I, III and IV of the ETC and, therefore, decreases oxidative phosphorylation. mDNA fragments are released from hepatocytes into blood and serve as a marker for early mitochondrial injury.
MPT
Protein nitration, such as nitrotyrosine formation, is a major stimulus for formation of the mitochondrial membrane permeability transition (MPT). The MPT describes formation of a pore in the inner membrane that allows molecules < 1500 Daltons to pass, which includes, of course, protons and other ions along with water. Development of the MPT is a common near final event in cell death in numerous pathological processes and can result from various stimuli, including an influx of Ca2+ into the matrix or, especially in this case, oxidant stress. That antioxidant scavengers of peroxynitrite, and that deferoxamine, which lessens nitrotyrosine formation by chelating iron, prevent or attenuate formation of the MPT speak to the importance of peroxynitrite as the major stimulus for MPT formation in APAP toxicity.
The exact proteins that change configuration and/or combine to create the MPT have not been definitely identified and may vary in different cell types. There is evidence for involvement of both the adenine nucleotide transporter as well as a subunit of ATP synthase. VDAC or other molecules (e.g. BAX) that won’t be discussed allow increased permeability in the outer membrane as well.
Transient MPT formation may normally occur during mitochondrial homeostasis. However, when the MPT is sustained, ions and water move into the matrix, causing mitochondrial swelling, which ruptures the outer mitochondrial membrane (Figure 17). This allows for the release of intermembrane proteins such as endonuclease G (Endo G) and apoptosis-inducing factor (AIF) into the cytosol. Endo G and AIF move into the nucleus where they catalyze nuclear DNA fragmentation. We now have reached the point of no return. Necrosis will ensue. Note that the time scale in Figure 17 represents the typical rapid progression of events in mice.
For those apoptosis fans, MPT formation in some disorders leads to the release of cytochrome C from the outside surface of the inner membrane into cytosol, which activates caspases, culminating in apoptosis. But evidence is overwhelming that necrosis, rather than apoptosis, characterizes hepatocyte death from APAP.

Adaptive Responses
In the face of oxidant stress, hepatocytes adapt in an attempt to reduce oxidants and maintain oxidative phosphorylation and mitochondrial integrity. The drop in inner membrane potential is accompanied by mitochondrial morphological changes of tubular mitochondria into biconcave discoid structures, which are initially reversible (Figure 18). This structural response may also result from influx of Ca2+ into the matrix. The discoid morphology and other responses such as contraction of cristae, increasing or decreasing the synthesis of ETC complexes, and changing expression of uncoupling proteins can affect the efficiency of oxidative phosphorylation.

Figure 18 – Rendering of a discoid mitochondrion as part of an adaptive response.
A recent paper, however, has addressed the opportunity to upregulate GSH synthesis. Another target of pJNK phosphorylation in the mouse turns out to be the catalytic subunit of glutamate-cysteine ligase (GCLC), the rate-limiting enzyme of the gamma-glutamyl cycle that is responsible for GSH synthesis. pJNK phosphorylates and inhibits GCLC, impairing restoration of GSH levels in hepatocytes and contributing to persistent JNK activation and oxidative stress.
Human Liver Cell Cultures and Patients with APAP Toxicity
Fresh cell cultures of human liver have been bathed in APAP solutions (and other substances), and events have been followed over time (Figure 19). Such events appear amazingly similar to those in mice, except that the time course is more prolonged, with necrosis appearing at about 24 hours. Even though GSH is depleted within 3 hours (30 min in mice), NAC is completely protective as long as provided within 6 hours, which speaks to the importance of preventing necrosis by restoring GSH. NAC definitely increases GSH synthesis, which neutralizes oxidant stress, thus interrupting the amplified oxidative/nitrosative stress cycle responsible for persistent JNK activation. Inhibition of JNK with SP600125, as in mice, attenuates cell death as well. As noted in the introduction, the course of events in cell cultures may be accelerated because of incubation in room air with a higher pO2 than that in the hepatic lobule.

Data from observational studies in human subjects with known APAP toxicity has allowed for measurements of various markers over time in blood (Figure 19). As in mice, plasma mDNA concentrations and glutamate dehydrogenase (GDH) activity (a mitochondrial enzyme) rise, reflecting mitochondrial injury. Both mice and human subjects show increases in circulating nuclear DNA fragments. Mice and humans share rises in plasma levels of similar damage-associated membrane proteins (DAMPs) from liver injury. Elevated circulating concentrations of APAP-protein adducts are seen in mice and man. Finally, circulating caspase markers of activated apoptosis are absent in both humans and mice.
Gradient of Injury
A gradient of hepatic injury exists during APAP toxicity, depending on the time following ingestion, dose, and timing of NAC therapy (Figure 20). For example, in the liver affected by NAPQI, severity of injury may range from full necrosis in hepatocytes immediately around the central vein, to cells in the midzonal region that still survive, but have undergone adaptive responses as they continue to experience persistent JNK activation and oxidant stress, but have not yet reached an irreversible sustained MPT, to periportal hepatocytes that are minimally affected or undergoing successful repair. The surviving hepatocytes are those that will give rise to cells for hepatic regeneration. While repair and regeneration compose a topic in and of itself and worthy of another post, the point is that it would be desirable to seek new therapeutic agents that may interrupt the amplified oxidative/nitrosative cycle before necrosis is established in surviving hepatocytes so that they may recover, including through removal or repair of damaged mitochondria.

Fomepizole
Physicians regularly give fomepizole to inhibit alcohol dehydrogenase in treatment of methanol and ethylene glycol poisoning and recognize it as a competitive inhibitor of CYP2E1. Indeed, Kang reported that human volunteers who ingested supratherapeutic doses of APAP (80 mg/kg) excreted dramatically lower amounts of APAP oxidative (CYP450) metabolites in urine when treated with fomepizole (Figure 21).

In 2019, Akakpo and colleagues reported protection of mice against toxic doses of APAP if fomepizole was given prior to APAP metabolism, with prevention of formation of APAP-protein adducts, increases in ROS, JNK activation, and necrosis, as would be expected from inhibition of CYP2E1. These results would also be similar to outcomes from giving NAC before or shortly after APAP dosing, but by maintaining GSH levels to detoxify NAPQI, rather than preventing NAPQI formation.
But the investigators did not stop there. They wondered what would happen if fomepizole was given 90 minutes after APAP dosing, which is well past the APAP metabolism stage in mice (Figure 22). At the time of this late fomepizole dosing, NAPQI already had depleted glutathione and formed protein adducts, complex III had begun creating superoxide, ASK1/2 had already been activated, MKK4 had been phosphorylated, and JNK activated to pJNK. But, pJNK had not yet translocated to the outer mitochondrial membrane, so had not yet phosphorylated Sab. Rather than observing increasing and persistent JNK activation (persistent pJNK), rising ROS levels, peroxynitrite formation, and release of Endo G and AIF, instead they found pJNK decreased over time to undetectable levels, and hepatic necrosis was prevented (Figure 22).

Fomepizole had interrupted the amplified oxidative stress cycle, but how? It couldn’t be by inhibiting CYP2E1. Interruption anywhere along the cycle could be responsible (Figure 23).

Molecular docking modeling using human pJNK (similar to mice) predicted that fomepizole bound to the ATP binding pocket in the active site of pJNK, preventing pJNK from phosphorylating substrates (Figure 24). And the scientists demonstrated fomepizole’s inhibition of pJNK’s kinase activity in in vitro assays and in other biological systems.

These results suggested that by preventing Sab phosphorylation, fomepizole interrupted the amplified oxidative stress cycle, stopped ROS production and halted activation of ASK1/2 (Figure 23). Presumably, MAKP phosphatases finally were able to prevail and dephosphorylate MKK4 and pJNK and prevent further JNK activation. GSH levels would rise again. Thus, statements such as “fomepizole inhibits JNK activation,” do not mean that fomepizole necessarily inhibits MKK4 to prevent phosphorylation and activation of JNK. Rather, current data suggest fomepizole inhibits the kinase activity of pJNK, itself, stopping the phosphorylation of Sab, and interrupting the amplified cycle, eventually leading to restoration of inactivated JNK. Importantly, the possibilities that fomepizole may also inhibit MKK4 or act elsewhere in the cycle have not been thoroughly examined, and additional actions may be found. For example, another competitive ATP antagonist, SP600125, inhibits both pJNK and MKK4, and so might fomepizole. So keep on the lookout for new publications adding to knowledge on this topic.
Akakpo and others returned in 2021 with a paper that described the effect of late fomepizole dosing in human hepatocytes. Hepatocytes were treated with APAP at time zero, with necrosis expected to appear at about 24 hours. But 18 hours after APAP dosing, cells were treated with NAC or fomepizole. Percent cell death was measured at 48 hours. Figure 25 shows low dose NAC had no effect, while high dose NAC did increase cell survival some. But fomepizole at low and high doses significantly outperformed NAC.

It can be difficult to extrapolate from drug doses that are effective in cell culture studies to doses used clinically, as drugs can concentrate in liver to achieve levels many fold greater than those in blood. But we can observe that the modeling studies of fomepizole’s binding to the active site of human pJNK predict a binding affinity that is slightly lower than that of its binding to the active site of human CYP2E1. And antagonism of CYP2E1 is easily achieved with 10 or 15 mg/kg fomepizole, doses used for treatment of toxic alcohol poisoning and as demonstrated in Kang’s study on APAP metabolism. Furthermore, standard extrapolation of the fomepizole dose that prevents JNK activation in mice to humans would suggest a human equivalent dose of about 4 mg/kg, more than 3 times lower than the standard 15 mg/kg loading dose used clinically.
Recall the concept of the gradient of injury, above. There may be areas in the liver where the late-acting effects of fomepizole are ineffective – cells are already committed to necrosis. But at the same time and in the same lobule there may be cells with lesser injury, on the path to necrosis, but with hepatocytes that can be salvaged through interruption of the cycle of continued JNK activation and oxidative/nitrosative stress. Fomepizole offers great excitement with regard to treating patients who present many hours after APAP ingestion or with established and worsening liver injury. Clinical trials are needed to determine if and when it is effective in human beings. Such trials should be of high priority.
Conclusion
The results from aforementioned studies along with fomepizole’s very high safety and low adverse effect profile have led medical toxicologists to use fomepizole for treatment of selected APAP OD patients (also receiving NAC), both early and late in their courses of illnesses, but without the benefit of results from clinical trials examining effectiveness. Hopefully, this post will help physicians understand their justification for doing so. Some would argue that NAC therapy became a standard of care long before results from the first clinical trial (with historical controls) demonstrated effectiveness, and adverse effects from NAC are more likely than from fomepizole.
On the other hand, deferoxamine was mentioned as attenuating formation of the MPT in mice. And minocycline and doxycycline can block Fe2+ uptake into the matrix through the Ca2+/Fe2+ uniporter, also limiting MPT formation in murine models. Recommendations for using deferoxamine and tetracycline derivatives to treat APAP poisoning, however, are far more distant in the future than the use of fomepizole, given that the potential for harm/adverse effects is greater. As examples, too much minocycline is toxic to mitochondria, and deferoxamine decreases renal blood flow. For now, clinical trials with fomepizole should be the priority.
Topics for additional individual posts on APAP toxicity could include mitochondrial adaptive responses, mitochondrial fission/fusion and mitophagy in recovery, the role of inflammation and cytokines in hepatocyte regeneration, effects of antidotes on hepatocyte regeneration, new strategies for treating APAP-induced liver injury, endothelial injury, Kupffer cells, and many more. Perhaps another day or another writer.
Postscript
Here are three discussion items for fellows to enrich their lives and keep them awake at night. Don’t worry. Your faculty members should have all the answers.
- Historically, what presumed amount of APAP taken in overdose was the oral NAC dosing regimen designed to address? Why might fomepizole given early assist in treatment of patients who take much larger amounts? And how were IV NAC dosing regimens chosen?
- A patient has just accidentally received a large overdose of IV APAP as a single bolus. Contrast the times of events in mice to your estimated times of events in this patient with regard to:
- Duration of NAPQI formation
- Peak GSH depletion
- Onset of JNK activation
- Translocation of JNK to external mitochondrial membranes
- Mitochondrial DNA fragmentation
- Onset of MPT formation with release of AIF and Endo G
- Established necrosis
- Then repeat your estimates, but following a massive oral APAP ingestion. And then with early or late NAC therapy. And then with early or late fomepizole therapy. And then with both NAC and fomepizole. And now do it all over for different zones of the hepatic lobule. I think you can see why we need clinical trials.
Selected References
General summaries
1. Ramachandran A, Jaeschke H. A mitochondrial journey through acetaminophen hepatotoxicity. Food Chem Toxicol. 2020 Jun;140:111282. doi: 10.1016/j.fct.2020.111282. Epub 2020 Mar 21. PMID: 32209353; PMCID: PMC7254872.
2. McGill MR, Hinson JA. The development and hepatotoxicity of acetaminophen: reviewing over a century of progress. Drug Metab Rev. 2020 Nov;52(4):472-500. doi: 10.1080/03602532.2020.1832112. Epub 2020 Oct 14. PMID: 33103516; PMCID: PMC8427730.
3 Ramachandran A, Jaeschke H. Mitochondria in Acetaminophen-Induced Liver Injury and Recovery: A Concise Review. Livers. 2023 Jun;3(2):219-231. doi: 10.3390/livers3020014. Epub 2023 Apr 10. PMID: 37377765; PMCID: PMC10299745.
4. Ramachandran A, Jaeschke H. Acetaminophen Toxicity: Novel Insights Into Mechanisms and Future Perspectives. Gene Expr. 2018 Mar 21;18(1):19-30. doi: 10.3727/105221617X15084371374138. Epub 2017 Oct 20. PMID: 29054140; PMCID: PMC5885144.
Mitochondrial CYP2E1
5. Hartman JH, Miller GP, Meyer JN. Toxicological Implications of Mitochondrial Localization of CYP2E1. Toxicol Res (Camb). 2017;6(3):273-289. doi: 10.1039/C7TX00020K. Epub 2017 Mar 14. PMID: 28989700; PMCID: PMC5627779.
6. Šrejber M, Navrátilová V, Paloncýová M, Bazgier V, Berka K, Anzenbacher P, Otyepka M. Membrane-attached mammalian cytochromes P450: An overview of the membrane’s effects on structure, drug binding, and interactions with redox partners. J Inorg Biochem. 2018 Jun;183:117-136. doi: 10.1016/j.jinorgbio.2018.03.002. Epub 2018 Mar 5. PMID: 29653695.
Animal models and cell cultures in APAP toxicity
7. Jaeschke H, Adelusi OB, Akakpo JY, Nguyen NT, Sanchez-Guerrero G, Umbaugh DS, Ding WX, Ramachandran A. Recommendations for the use of the acetaminophen hepatotoxicity model for mechanistic studies and how to avoid common pitfalls. Acta Pharm Sin B. 2021 Dec;11(12):3740-3755. doi: 10.1016/j.apsb.2021.09.023. Epub 2021 Sep 30. PMID: 35024303; PMCID: PMC8727921.
Redox signaling
8. Collins Y, Chouchani ET, James AM, Menger KE, Cochemé HM, Murphy MP. Mitochondrial redox signalling at a glance. J Cell Sci. 2012 Feb 15;125(Pt 4):801-6. doi: 10.1242/jcs.098475. Erratum in: J Cell Sci. 2012 Apr 1;125(Pt 7):1837. PMID: 22448036.
9. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020 Jul;21(7):363-383. doi: 10.1038/s41580-020-0230-3. Epub 2020 Mar 30. PMID: 32231263.
ASK1 and APAP
10. Xie Y, Ramachandran A, Breckenridge DG, Liles JT, Lebofsky M, Farhood A, Jaeschke H. Inhibitor of apoptosis signal-regulating kinase 1 protects against acetaminophen-induced liver injury. Toxicol Appl Pharmacol. 2015 Jul 1;286(1):1-9. doi: 10.1016/j.taap.2015.03.019. Epub 2015 Mar 25. PMID: 25818599; PMCID: PMC4444402.
Superoxide and JNK activation
11. Nguyen NT, Du K, Akakpo JY, Umbaugh DS, Jaeschke H, Ramachandran A. Mitochondrial protein adduct and superoxide generation are prerequisites for early activation of c-jun N-terminal kinase within the cytosol after an acetaminophen overdose in mice. Toxicol Lett. 2021 Mar 1;338:21-31. doi: 10.1016/j.toxlet.2020.12.005. Epub 2020 Dec 5. PMID: 33290831; PMCID: PMC7852579.
Sab phosphorylation and Src inactivation
12. Win S, Than TA, Min RW, Aghajan M, Kaplowitz N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology. 2016 Jun;63(6):1987-2003. doi: 10.1002/hep.28486. Epub 2016 Mar 15. PMID: 26845758; PMCID: PMC4874901.
Lysosomal iron
13. Hu J, Kholmukhamedov A, Lindsey CC, Beeson CC, Jaeschke H, Lemasters JJ. Translocation of iron from lysosomes to mitochondria during acetaminophen-induced hepatocellular injury: Protection by starch-desferal and minocycline. Free Radic Biol Med. 2016 Aug;97:418-426. doi: 10.1016/j.freeradbiomed.2016.06.024. Epub 2016 Jun 23. PMID: 27345134; PMCID: PMC4996678.
14. Kon K, Kim JS, Uchiyama A, Jaeschke H, Lemasters JJ. Lysosomal iron mobilization and induction of the mitochondrial permeability transition in acetaminophen-induced toxicity to mouse hepatocytes. Toxicol Sci. 2010 Sep;117(1):101-8. doi: 10.1093/toxsci/kfq175. Epub 2010 Jun 27. PMID: 20584761; PMCID: PMC2923283.
Peroxynitrite formation
15. Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol Sci. 2001 Aug;62(2):212-20. doi: 10.1093/toxsci/62.2.212. PMID: 11452133.
Mitochondrial morphological changes
16. Umbaugh DS, Nguyen NT, Jaeschke H, Ramachandran A. Mitochondrial Membrane Potential Drives Early Change in Mitochondrial Morphology After Acetaminophen Exposure. Toxicol Sci. 2021 Feb 26;180(1):186-195. doi: 10.1093/toxsci/kfaa188. PMID: 33432343; PMCID: PMC7916734.
17. Ahmad T, Aggarwal K, Pattnaik B, Mukherjee S, Sethi T, Tiwari BK, Kumar M, Micheal A, Mabalirajan U, Ghosh B, Sinha Roy S, Agrawal A. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013 Jan 17;4(1):e461. doi: 10.1038/cddis.2012.213. PMID: 23328668; PMCID: PMC3564000.
Inhibition of JNK by fomepizole
18. Akakpo JY, Ramachandran A, Duan L, Schaich MA, Jaeschke MW, Freudenthal BD, Ding WX, Rumack BH, Jaeschke H. Delayed Treatment With 4-Methylpyrazole Protects Against Acetaminophen Hepatotoxicity in Mice by Inhibition of c-Jun n-Terminal Kinase. Toxicol Sci. 2019 Jul 1;170(1):57-68. doi: 10.1093/toxsci/kfz077. PMID: 30903181; PMCID: PMC6592188.
Lipid peroxidation in APAP toxicity
19. Jaeschke H, Ramachandran A. Oxidant Stress and Lipid Peroxidation in Acetaminophen Hepatotoxicity. React Oxyg Species (Apex). 2018 May;5(15):145-158. Epub 2018 May 1. PMID: 29682614; PMCID: PMC5903282.
Cell culture studies
20. Yan HM, Ramachandran A, Bajt ML, Lemasters JJ, Jaeschke H. The oxygen tension modulates acetaminophen-induced mitochondrial oxidant stress and cell injury in cultured hepatocytes. Toxicol Sci. 2010 Oct;117(2):515-23. doi: 10.1093/toxsci/kfq208. Epub 2010 Jul 8. PMID: 20616211; PMCID: PMC2940407.
21. McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H. HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology. 2011 Mar;53(3):974-82. doi: 10.1002/hep.24132. Epub 2011 Feb 11. PMID: 21319200; PMCID: PMC3073317.
22. Akakpo JY, Jaeschke MW, Ramachandran A, Curry SC, Rumack BH, Jaeschke H. Delayed administration of N-acetylcysteine blunts recovery after an acetaminophen overdose unlike 4-methylpyrazole. Arch Toxicol. 2021 Oct;95(10):3377-3391. doi: 10.1007/s00204-021-03142-9. Epub 2021 Aug 22. PMID: 34420083; PMCID: PMC8448936.
23. Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol. 2014 Sep 15;279(3):266-274. doi: 10.1016/j.taap.2014.05.010. Epub 2014 Jun 3. PMID: 24905542; PMCID: PMC4171351.
24. Halliwell B. Oxidative stress in cell culture: an under-appreciated problem? FEBS Lett. 2003 Apr 10;540(1-3):3-6. doi: 10.1016/s0014-5793(03)00235-7. PMID: 12681474.
JNK inhibition of GSH synthesis
24. Win S, Than TA, Kaplowitz N. c-Jun-N Terminal Kinase-Mediated Degradation of γ-Glutamylcysteine Ligase Catalytic Subunit Inhibits GSH Recovery After Acetaminophen Treatment: Role in Sustaining JNK Activation and Liver Injury. Antioxid Redox Signal. 2023 Jun;38(16-18):1071-1081. doi: 10.1089/ars.2022.0119. Epub 2023 Jan 5. PMID: 36333933.
Human overdose studies
25. McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012 Apr;122(4):1574-83. doi: 10.1172/JCI59755. Epub 2012 Mar 1. PMID: 22378043; PMCID: PMC3314460.
26. Curry SC, Padilla-Jones A, Ruha AM, O’Connor AD, Kang AM, Wilkins DG, Jaeschke H, Wilhelms K, Gerkin RD; Acetaminophen Adduct Study Group. The Relationship Between Circulating Acetaminophen-Protein Adduct Concentrations and Alanine Aminotransferase Activities in Patients With and Without Acetaminophen Overdose and Toxicity. J Med Toxicol. 2019 Jul;15(3):143-155. doi: 10.1007/s13181-019-00705-2. Epub 2019 Apr 12. PMID: 30980348; PMCID: PMC6597749.
27. Curry SC, Padilla-Jones A, O’Connor AD, Ruha AM, Bikin DS, Wilkins DG, Rollins DE, Slawson MH, Gerkin RD; Acetaminophen Adduct Study Group. Prolonged Acetaminophen-Protein Adduct Elimination During Renal Failure, Lack of Adduct Removal by Hemodiafiltration, and Urinary Adduct Concentrations After Acetaminophen Overdose. J Med Toxicol. 2015 Jun;11(2):169-78. doi: 10.1007/s13181-014-0431-2. PMID: 25288219; PMCID: PMC4469721.
Fomepizole and APAP metabolism in humans
28. Kang AM, Padilla-Jones A, Fisher ES, Akakpo JY, Jaeschke H, Rumack BH, Gerkin RD, Curry SC. The Effect of 4-Methylpyrazole on Oxidative Metabolism of Acetaminophen in Human Volunteers. J Med Toxicol. 2020 Apr;16(2):169-176. doi: 10.1007/s13181-019-00740-z. Epub 2019 Nov 25. PMID: 31768936; PMCID: PMC7099124.
Comparing NAC and fomepizole as APAP antidotes
29. Akakpo JY, Ramachandran A, Curry SC, Rumack BH, Jaeschke H. Comparing N-acetylcysteine and 4-methylpyrazole as antidotes for acetaminophen overdose. Arch Toxicol. 2022 Feb;96(2):453-465. doi: 10.1007/s00204-021-03211-z. Epub 2022 Jan 3. PMID: 34978586; PMCID: PMC8837711.
Mouse APAP-protein adducts
30. McGill MR, Lebofsky M, Norris HR, Slawson MH, Bajt ML, Xie Y, Williams CD, Wilkins DG, Rollins DE, Jaeschke H. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose-response, mechanisms, and clinical implications. Toxicol Appl Pharmacol. 2013 Jun 15;269(3):240-9. doi: 10.1016/j.taap.2013.03.026. Epub 2013 Apr 6. PMID: 23571099; PMCID: PMC3654056.
Fomepizole as novel strategy
31. Jaeschke H, Akakpo JY, Umbaugh DS, Ramachandran A. Novel Therapeutic Approaches Against Acetaminophen-induced Liver Injury and Acute Liver Failure. Toxicol Sci. 2020 Apr 1;174(2):159-167. doi: 10.1093/toxsci/kfaa002. PMID: 31926003; PMCID: PMC7098369.
This was excellent!!
Is there any evidence of harm in giving fomepizole late? When would you guess too late would be in people?
Is there any evidence that giving NAC with fomepizole late would be a problem?
Is there any evidence that giving fomepizole early (with APAP concentration above ?900 mcg/mL and a metabolic acidosis) with or without NAC would be detrimental? Does APAP itself play any part in the development of the metabolic acidosis or is it just NAPQI?
Thank you!! This was amazing and truly translational between the basic sciences and clinical toxicology!!