, 1893. Public Domain from wikimedia.org](https://i0.wp.com/toxandhound.com/wp-content/uploads/2020/07/hero-image.png?resize=982%2C526&ssl=1)
{kind=link}
Whence the Protons of Lactic Acidosis?
The case of impaired mitochondrial electron transport
Steven C Curry, MD
University of Arizona College of Medicine – Phoenix
Banner – University Medical Center Phoenix
Phoenix, AZ
@SteveCurryMD
Robert A Robergs, Ph.D.
School of Exercise and Nutrition Sciences, Faculty of Health
Queensland University of Technology
Brisbane, QLD, Australia
**NEW** – When finished with this first part, please see the update – Part II – Cellular Efflux of Protons and Lactate.
Introduction
Last year I wrote posts on various topics of my choosing, but for the first time Howard Greller asked me to address a specific topic – the source of protons in so-called lactic acidosis. Asking from where protons arise during lactic acidosis may sound rhetorical, but this is hardly the case. My interest in the topic began in 1974 during my first biochemistry course. I was told that during lack of oxygen, pyruvate was converted to lactate, and accumulation of lactic acid produced a severe acidosis. When I pointed out that pyruvate and lactate were almost 100% ionized at physiologic pH and the book showed that conversion of pyruvate to lactate actually consumed a proton, my professor could offer no explanation and simply referred me back to the book, which continued to illustrate my point:

The name “lactic acidosis”, unfortunately, reinforces the common misconception that protons responsible for metabolic acidosis have dissociated from lactate, even though it has been recognized for about a half century this is not the case. Even Krebs noted that conversion of glucose to lactate was not acidifying. That lactate is not responsible for metabolic acidosis explains several important observations in medicine which we have taught in our medical toxicology fellowship in Phoenix for more than 35 years.
Thus, I agreed to Howard’s request, but brought along Rob Robergs, Ph.D. as a contributor to this post. Rob is probably the world’s top expert on proton balance in glycolysis and related pathways. I have been reading his papers for years and have communicated with him for some time. He has been kind enough to accept my invitation. For the purpose of this post, we will be consistent with most authorities and consider glycolysis to represent the cytoplasmic anaerobic conversion of glucose to pyruvate, rather than glucose to lactate. I am restricting this column to proton balance during lactic acidosis resulting from impaired mitochondrial electron transport, such as from poisoning by various toxins, hypoxia, or severe tissue hypoperfusion. As in previous columns, things must remain simplified because of length limitations and short attention spans of readers, but I’ve supplied references for those who desire more detail. And feel free to fire away with questions.
Background
Before we start tracking protons, we need to briefly review some basic biochemistry of glycolysis and oxidative phosphorylation to remind us of what we once learned. Figure 2 superficially summarizes the complete oxidation of glucose that was beaten into our brains in college and medical school.

In the cytoplasm, glucose is converted to 2 pyruvate molecules via glycolysis. Pyruvate then moves into the matrix of the mitochondrion where it is converted to acetyl-CoA and combines with oxaloacetate to enter the tricarboxylic acid (TCA) cycle, more commonly known as the Krebs cycle (or citric acid cycle). NADH and FADH produced in the Krebs cycle donate electrons to the electron transport chain (ETC) in the inner mitochondrial membrane (cristae), where they shuttle between various complexes before being placed on oxygen, the terminal electron acceptor, to form H2O. Protons are used to create an electrochemical gradient that fuels ATP synthesis which I describe later. Krebs cycle also produces a GTP which is converted to ATP for each cycle.
Glycolysis is anaerobic. ATP production in mitochondria requires oxygen and, thus, represents oxidative phosphorylation. While glycolysis produces a net 2 ATP (net 3 ATP when starting with glycogen), the addition of oxidative phosphorylation allows net production of 38 ATP from each glucose molecule.
Most of us have been taught at one time or another that during impaired oxygen delivery to tissue, whether from hypoxia and/or hypoperfusion, oxidative phosphorylation stops and we rely on glycolytic production of ATP, alone. In order to regenerate adequate quantities of NAD+ from NADH, pyruvate is reduced to lactate with consumption of a proton (Figure 3). The conversion of pyruvate to lactate easily allows regeneration of NAD+ and restoration of a normal redox potential (reflected in an NADH/NAD+ ratio) in many situations in which NAD+ has been consumed, such as ethanol metabolism (discussed below).

The overall reaction of glycolysis is written as:

Take a look at how phosphate (Pi), AMP, ADP and ATP allosterically modulate the rate of glycolysis (Figure 5). When Pi, AMP or ADP levels rise (from ATP hydrolysis), glycolysis accelerates, increasing ATP and pyruvate production. We can really rev up glycolysis and pyruvate production when demand for ATP rises, though the inefficiency of glycolytic ATP synthesis cannot sustain life for long except in specialized cells such as erythrocytes, which have a low ATP demand.

Hyperlactatemia
Simplistically, we can consider three main reasons patients experience rises in circulating lactate concentrations: increased production; decreased clearance; and changes in redox potential.
Increased production
Increased glycolytic pyruvate production with or without impaired oxidative phosphorylation can, understandably, raise lactate levels. Increased production during intense exercise serves as an example.
Decreased clearance
Normally lactate is taken up by the liver and kidneys where it is oxidized back to pyruvate and subsequently converted back to glucose-6-phosphate (Cori cycle). However, during times of systemic impairment of oxidative phosphorylation (or with hepatic ischemia), the liver becomes a lactate producer. Interestingly, the liver produces lactate only when there is adequate glycogen. Glycogenolysis to glucose-6-phosphate and then through glycolysis is the path by which the liver forms lactate. Lactate is filtered by the kidneys, but almost all is reabsorbed in the proximal tubule at normal lactate concentrations.
Changes in redox potential
There exists an equilibrium between pyruvate and lactate. Normal plasma lactate levels are about 10 times higher than pyruvate concentrations, with a lactate concentration of about 1 mM (1 mEq/L). This baseline circulating lactate mainly comes from erythrocytes, which have no mitochondria and survive on glycolysis and the hexose monophosphate shunt, alone.
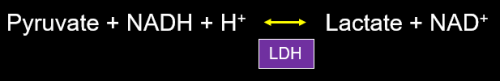
The equilibrium constant, K, for this reaction is the product of the molar concentrations of products on the right divided by reactants on the left. Using a bit of algebra we arrive at the following:

The higher the NADH/NAD+ ratio, the higher the lactate/pyruvate ratio. Apart from rates of lactate production or clearance, a change in the NADH/NAD+ ratio, itself, will be associated with a raised or lowered lactate concentration. Let’s take the case of ethanol. The oxidative metabolism of ethanol through alcohol dehydrogenase results in the conversion of NAD+ to NADH, raising the NADH/NAD+ ratio. Persons with blood alcohol concentrations of 100 mg/dL (0.1%) commonly develop plasma lactate concentrations of about 5 mM.

The ingestion of a large meal will also raise postprandial lactate levels from increased glycolysis and pyruvate formation. Increased beta oxidation of fatty acids (lipolysis) during ketoacidosis also generates large amounts of NADH, raising the lactate to pyruvate ratio.
When the lactate levels rise only as a result of changes in redox potential in everyday life, they generally don’t exceed about 5 to 7 mM in the face of normal kidney function, at least partly because the threshold for renal tubular reabsorption of filtered lactate is about 6.6 mM, and urinary excretion increases at higher lactate concentrations.
The Protons of Glucose to Lactate
In glycolysis protons could arise from three major sources: 1) reduction of NAD+ to form NADH + H+; 2) dissociation from a phosphate moiety; or 3) dissociation from a carboxylic acid group.
Remember that hydrogen is only an electron and a proton. During oxidation reactions, 2 electrons removed from a molecule along with 1 proton combine with NAD+ to form NADH, with one proton left over: NAD+ → NADH + H+.
Phosphate groups are completely or partially ionized at physiologic pH. Pure ATP in water is actually a quadruple acid with 4 dissociable protons:

In vivo, ATP is always bound to a Mg2+ molecule, making it a divalent anion with two dissociable protons:

Similarly, ADP, always bound to Mg2+, is an anion with 1 dissociable proton:

Inorganic phosphate (Pi) in water can dissociate between 1 (H2PO4– + H+) and 2 protons (HPO42- + 2 H+) at pH values consistent with life.
ATP hydrolysis generates a proton. We start off with 2 protons and end up with 3, with a net increase of one proton for each ATP hydrolyzed. In Figure 12, I will include the Mg, but this is not normally done by convention.

With regard to carboxylic acid groups, take a look at the simplified figure of glycolysis, below. Read left-to-right like a book. We begin with glucose and end with pyruvate on the bottom right.
](https://i0.wp.com/toxandhound.com/wp-content/uploads/2020/07/figure-13.png?resize=750%2C439&ssl=1)
Note that the first carboxylic group to appear is 3-phosphoglycerate at the right end of the middle portion of the figure. That single carboxylic group remains unchanged while the rest of the molecule undergoes sequential modifications to eventually become pyruvate. The figure (from Wikipedia) suggests that protons from the carboxylic acid groups of 3-phosphoglycerate, 2-phosphoglycerate and phosphoenolpyruvate are not dissociated, but this is not the case. When a phosphate group is removed in the synthesis of 3-phosphoglycerate, the remaining oxygen remains unbound and negatively charged at physiologic pH, causing the nomenclature of the product to be an ion (‘ate’ at the end of the name). At a pH compatible with life, the great majority of these so-called tricarboxylic acids are produced in their dissociated forms, given their low pKa values. We do not create meaningful amounts of carboxylic acid groups with newly dissociated protons as 1,3-biphosphoglycerate is sequentially converted to the pyruvate anion.
Basic Proton Balance of Glucose to Lactate
The table below (Figure 14) shows the reactions of glycolysis as commonly presented in biochemistry texts, which are without adjustment for pH-dependent partial ionization. Let’s use these assumptions in our first effort to follow protons.
During glycolysis a total of 4 protons are released, and 2 protons are consumed, resulting in the net formation of 2 protons and 2 pyruvate. Importantly, none of these protons arise from carboxylic acid groups. In fact, in the last step of glycolysis, the formation of 2 pyruvate actually consumes, rather than creates, 2 protons. The single step of glycolysis that releases the greatest number of protons is the formation of 1,3-biphosphoglycerate, which is not an acid.

Thus, glycolysis, alone, is acidifying, but protons do not come from pyruvate. The net reaction of glycolysis, once again, is:

What happens when we convert 2 pyruvate to 2 lactate in order to regenerate NAD+? We consume yet another 2 protons (Figure 16), giving us a net proton balance for glucose to lactate of zero (Figure 14).

Thus, glucose to lactate is not acidifying. In fact, we never created pyruvic or lactic acid. Rather, we created the pyruvate and lactate anions.

For those of you compulsive about ions and who want to assume that protons are the only cations around, we begin and end glucose to lactate with the same number of protons. This can be demonstrated with and without considering Mg2+. Both of the following equations represent the same reaction as in Figure 17:

Before we move on to more complicated proton balancing at a lower pH, you should be asking yourselves where in the world the protons come from when lactate levels rise during “lactic acidosis”. Krebs pointed out that the daily turnover of H+ ions is greater than any other intermediary metabolite, and this is “because the most frequent single reaction in aerobic cells is the conversion of ATP to ADP and Pi, which at the pH of the cell is accompanied by a stoichiometric formation of H+ ions”. Once again, ATP hydrolysis is acidifying (Figure 12), with the net reaction being:

When the ATP generated in glycolysis undergoes hydrolysis, protons are released, as illustrated below.

When considering that an ATP formed in glycolysis is quickly hydrolyzed to release a proton, we arrive at the commonly written equation in Figure 21, which implies that glucose forms lactic acid. In reality, lactate comes from glucose, while protons come from hydrolysis of ATP in an unrelated reaction. Glucose to lactate generates 2 lactate anions and no protons. Only after hydrolysis of ATP in a separate reaction, apart from lactate synthesis, do protons appear, though this occurs almost instantaneously.

This explains, for example, why in their 2014 review of lactic acidosis in the New England Journal of Medicine, Kraut and Madias wrote that:
. . . production of lactate ions by means of glycolysis is accompanied by the release
of an equivalent number of protons from the hydrolysis of the generated ATP.
They got it right – glycolysis produces lactate anions, and ATP hydrolysis produces protons. Many authors have been pointing this out for about 50 years, with important contributions by Rob, our guest contributor. We certainly have been teaching it at our medical toxicology fellowship since the 1980s. Unfortunately, many texts and scientific papers continue to claim, incorrectly, that lactic acid is formed in glycolysis and accumulates to produce a metabolic acidosis during times of impaired oxidative phosphorylation. We will examine some implications of the disconnect between lactate formation and metabolic acidosis later.
Protons and Glycolytic ATP synthesis
The next question you may ask is why doesn’t glycolytic synthesis of ATP via reversal of the ATP hydrolysis reaction simply consume the total of 4 protons resulting from glycolysis and 2 ATP hydrolysis? Take a look at Figure 22, below, and note the red arrows. ATP is synthesized in two steps, and in neither step does ATP synthesis result from the reverse of its hydrolysis. When 2 ATP are synthesized along with 3-phosphoglycerate, no protons are consumed. When 2 ATP are synthesized with pyruvate, 2 protons, indeed, are consumed, but not by reversal of ATP hydrolysis. Regardless, glycolytic ATP synthesis cannot consume all the protons generated during glycolysis and resulting from ATP hydrolysis.

Protons and Oxidative Phosphorylation
We have seen that glycolytic oxidation of glucose to 2 pyruvate is acidifying, with generation of 2 protons. In normal conditions most pyruvate is transported into the mitochondrial matrix to fuel the Krebs cycle and oxidative phosphorylation rather than reduced to lactate. Why, then, does glycolysis, which exists in virtually every cell, not generate a metabolic acidosis in everyone? After all, both glycolysis and the hydrolysis of the ATP it generates produce protons.
To understand this, we first must quickly take a look at simplified oxidative phosphorylation. We have already examined oxidative phosphorylation, especially cytochrome oxidase, in somewhat more detail in the post on cyanomythology, and have looked into additional mitochondrial functions when discussing hepatic microvesicular steatosis. This will be simpler.
A section of the cristae is shown in Figure 23. The matrix is on the bottom, and the intermembrane space, which communicates with cytoplasm, is on top.

Electrons from NADH are shuttled onto complex I of the electron transport chain (ETC). Complex I then shuttles electrons to ubiquinone (Q), then to complex III, then to cytochrome C on the external surface of the inner mitochondrial membrane, and finally onto complex IV, better known as cytochrome oxidase, where they combine with oxygen to make water. In the Krebs cycle, the oxidation of succinate results in formation of FADH2 which donates electrons to complex II. These electrons also move to Q before eventually making their way to oxygen.
The energy released from electrons moving down the ETC is used to pump protons from the matrix into the intermembrane space through complexes I, III and IV. The protons then reenter the matrix down their electrochemical gradient through ATP synthase, and this energy is used to make ATP. Not shown in the figure are countless transporters/exchangers in the inner membrane, including those for ATP, ADP and Pi.
Note that Figure 23 illustrates that ATP synthase generates ATP by reversal of the ATP hydrolysis reaction. In fact, ATP synthase is actually an ATPase. In the absence of the large proton gradient across the inner mitochondrial membrane, this enzyme would hydrolyze ATP and move protons out of the matrix, into the intermembrane space. The much higher proton concentration in the intermembrane space than in the matrix created by electron transport forces protons backwards through ATP synthase, which then runs the hydrolysis reaction in reverse, synthesizing ATP and consuming protons. When the inner membrane proton gradient collapses, such as during times of uncoupling of oxidative phosphorylation, ATP synthase returns to becoming an ATPase and actually further depletes ATP stores and adds to production of metabolic acidosis via ATP hydrolysis.
But proton movement into and out of the matrix is much more complicated. Below is a very oversimplified illustration of a fraction of the various mechanisms by which protons move across the mitochondrial inner membrane. Apart from complexes I, III and IV and ATP synthase, there are various ion transporters that involve protons – the sodium-proton exchanger is shown as a representative of all of them. The phosphate transporter (blue) moves both phosphate and a proton into the matrix. There is a proton leak across the inner membrane that accounts for a significant portion of our baseline oxygen consumption. Not shown are uncoupling proteins and transporters for various organic compounds, including some intermediates of the Krebs cycle. And much more. With all of this going on, how can we determine whether oxidative phosphorylation consumes or generates net protons?

This was a question addressed by Dr. Pál Váhy in 1979.

Very briefly, Dr. Váhy prepared mitochondrial suspensions and measured oxygen consumption and extramitochondrial proton concentrations (pH). He found that increased mitochondrial respiration, measured as an increase in oxygen consumption, was accompanied by a fall in extramitochondrial protons (rise in pH), as they moved into the matrix. Importantly, when mitochondrial respiration stopped, the protons remained in the mitochondria, indicating their movement was not simply a transient influx, but represented real consumption. When examining the stoichiometry of proton generation and mitochondrial uptake, Dr. Váhy’s in vitro studies strongly suggested that oxidative phosphorylation was able to counterbalance the cytoplasmic proton-generating processes of ATP hydrolysis. I crudely created a figure summarizing his findings, below.

Various toxins inhibit electron transport and shut down oxidative phosphorylation. As examples, cyanide (HCN), azide (HN3), sulfide (H2S), and formic acid (including that from methanol metabolism) combine to the binuclear center of cytochrome a3 on complex IV to stop electron transport, oxygen consumption and, therefore, ATP production. Metformin inhibits at complex I and rotenone prevents transfer of electrons from complex I to Q at toxic concentrations. Poisonings by all of these agents are accompanied by increased glycolytic production of pyruvate which, in turn, is converted to lactate, along with a metabolic acidosis. And the same can be said for asphyxiants which displace oxygen in the atmosphere. Of course, any cause of hypoxemia or impaired O2 delivery can do the same.
Here’s the big picture, then. Normally, pyruvate generated in glycolysis moves into mitochondria where it fuels Krebs cycle and oxidative phosphorylation (Figure 27). Protons from the hydrolysis of ATP generated in glycolysis are taken up by mitochondria and consumed during oxidative phosphorylation, maintaining pH.

Except for forming some creatine phosphate, we do not store ATP. We maintain a steady ATP/ADP ratio in cells by synthesizing ATP at the same rate we consume it. And synthesizing ATP in oxidative phosphorylation is our major buffer for proton production. Krebs pointed out that our daily adult ATP production is on the order of 62 Kg, consuming about 150,000 mmoles of protons. Of course, we daily hydrolyze that same amount of ATP and generate as many protons, keeping everything in balance. Certainly some protons from glycolysis and ATP hydrolysis are buffered by bicarbonate, undergo urinary excretion (e.g., ammonium excretion), or are consumed in various other reactions. But these processes are relatively minor compared to the truly massive consumption of protons by oxidative phosphorylation, the topic we are focusing on.
The consequences of impaired electron transport and oxidative phosphorylation are shown in Figure 28. Glycolysis accelerates with increased production of pyruvate, which goes to lactate without net production of protons. The hydrolysis of ATP created in glycolysis generates protons, but those protons now cannot be buffered by oxidative phosphorylation. Thus, circulating lactate levels rise and we develop a metabolic acidosis that is known by the misnomer, lactic acidosis.

Adjusted Proton Balance
Some of you are thinking that there are lots of factors that influence dissociation of protons from substrates of glycolysis. Certainly, proton dissociation/binding to intermediates changes with pH. And protons must compete for binding with other intracellular cations, mainly Na+, K+, Mg2+ and Ca2+, to the intermediates and products of glycolysis. These cations are present at concentrations about 10,000 to 1 million times greater than those of protons, though the small size of protons gives them a binding advantage. These and other factors are why I invited Rob to join me in this column.
Rob has focused years of research on taking into consideration numerous parameters in complicated analyses of the proton balance of glycolysis and of glucose to lactate at different pH values, and with adjustments for other factors, including competing cations, partial ionization (dissociation) of protons, and flux of substrates in glycolysis. As an example, below is a table of proton balance for the reactions of glycolysis at pH 7 in skeletal muscle.

When all reactions are considered, the conversion of glucose to 2 lactate is slightly acidifying at pH 7, with generation of about 0.67 moles of protons. But again, none of the protons come from lactate. In fact, as pH declines, more protons bind to lactate, with lactate serving as a base and buffer. The same is true for pyruvate.
We must now consider what the lower pH has done to the proton balance of ATP hydrolysis, shown below:

The low pH drives protons onto phosphate, increasing the fraction that exists as H2PO4– rather than HPO42-, resulting in fewer dissociated protons.
Thus, when we add the protons from conversion of glucose to 2 lactate (0.6727) to the protons from 2 ATP hydrolysis (1.327), we get 1.9997 protons, which is virtually unchanged from our earlier example. Again, equations showing glucose going to 2 lactate and 2 protons do not represent glycolytic conversion of glucose to pyruvate followed by reduction to lactatic acid. Rather such an equation shows what we are left with after taking glucose to lactate and then hydrolyzing the ATP in a separate reaction. Summarizing at pH 7:

So, at pH 7 when adjusting for partial proton dissociation, competitive binding by other cations, intracellular substrate concentrations and other factors, most protons still arise from ATP hydrolysis, though a fraction do arise from glycolysis. But even those protons do not come from lactate. They arise from formation of “NADH + H+” and from phosphates. Lactate is never a meaningful source of protons in metabolic acidosis at pH values compatible with life.
Implications and Conclusions
Why should you care whether the protons responsible for metabolic acidosis during times of hyperlactatemia come from ATP and, at low pH, from dissociation of other intermediates of glycolysis, rather than from lactic acid? Is this something that should keep you awake at night? Based on arguments I have read on web sites and heard on podcasts, some persons become extremely upset when someone claims that lactic acid is not responsible for metabolic acidosis. Let us pray for their wisdom and peace of mind.
The dissociation between lactate concentrations and proton formation responsible for metabolic acidosis explains various observations that can assist us clinically. I’ll list a few below, in no particular order:
- Lactate levels can rise in the absence of a metabolic acidosis from impaired oxidative phosphorylation. We certainly see this with changes in redox potential after eating, consuming alcohol, in ketoacidosis, starvation, and in other states. We also see it with increased pyruvate levels and its conversion to lactate with a normal redox potential. As long as oxidative phosphorylation is working well to buffer protons, pH will remain normal unless an acid/base disturbance results from another etiology.
- When metabolic acidosis is present, the base deficit frequently does not correlate well with lactate concentration.
- During metabolic acidosis from impaired electron transport and oxidative phosphorylation, increases in anion gaps usually do not match increases in lactate concentrations.This is the rule, not the exception. You may have a patient in shock with pH 6.9, an anion gap of 37 mEq/L and an arterial plasma lactate concentration of 10 mEq/L (mM). The Δ gap of about 22 cannot be entirely explained by an increase in lactate to 10. And, of course, it shouldn’t, since none of the protons responsible for acidosis that are being offset by unmeasured anions came from lactic acid.
- We have been restricting today’s topic to impaired electron transport. But how about uncouplers of oxidative phosphorylation? During uncoupling (e.g., salicylate or nitrophenol poisoning), when electron transport and oxygen consumption are accelerated, oxidative phosphorylation is impaired and glycolysis increases in an attempt to support cellular ATP needs. During uncoupling, the NADH/NAD+ ratio can decrease, since the accelerated electron transport is fueled mainly by NADH being converted to NAD+. A decreased NADH/NAD+ ratio results in a decreased lactate/pyruvate ratio with a lower lactate level for a given pyruvate concentration. I have commonly found normal or minimally elevated lactate levels in patients quite ill from salicylate toxicity. (Salicylate also inhibits dehydrogenases in Krebs cycle.)
None of our discussion today goes against the value of measuring plasma lactate concentrations. There is no doubt they can be of prognostic significance and provide support for diagnostic suspicion of intestinal ischemia, beriberi, sepsis, and other disorders. But lactic acid is not responsible for any component of metabolic acidosis during times of impaired oxidative phosphorylation, shock, asphyxia or hypoxemia. Lactate levels are reflections of rates of lactate formation (or infusion of lactate salts), redox potential, lactate clearance by liver and kidney in the Cori cycle, and lactate clearance via urinary excretion, especially when the tubular reabsorption threshold is exceeded. Toss in lactate removal by hemodialysis or CRRT, too.
As we finish up, it’s best to remain calm. It’s perfectly fine and normal that lactic acid does not produce “lactic acidosis”. Instead, lactate anions buffer protons and lessen the decline in pH as acidemia worsens. It’s just something you will need to live with.
Feel free to leave questions, and please, please leave feedback. I need to understand what sorts and depth of topics readers would like to read.
Postscript
I offer a few questions for medical toxicology fellows to consider and discuss with faculty members to explore the implications of what was discussed today.
- Assume we have identical twins who are in excellent health, of identical weight, and taking no medications. One overdoses with 2,4-dinitrophenol to impair oxidative phosphorylation by 30% (just tossing a number out there). The second twin overdoses with sodium cyanide to impair oxidative phosphorylation by 30%. Both have maintained identical renal function. Which one is likely to have the higher lactate level and why? You may want to consider changes in NADH/NAD+ ratios with different types of inhibition of oxidative phosphorylation. A review of the post on hepatic microvesicular steatosis may assist you.
- Figures 7 shows that the NADH/NAD+ ratio is proportional to the lactate/pyruvate ratio. If you follow the algebra in the figure, you may have noted that the proton was dropped. We could also write what is in Figure 32, below:

This implies that a rise in proton concentration (drop in pH), alone, should be associated with raised lactate concentrations, regardless of etiology. But this is believed to be a rather unimportant and minor influence. That is, lowering pH, alone, without a change in the NADH/NAD+ ratio, is not thought to be associated with a significant rise in the lactate/pyruvate ratio. What is the effect of a drop in pH, alone, on phosphofructokinase and what effect, in turn, would this have on pyruvate and lactate production? In fact, what does hyperventilation and creation of a respiratory alkalosis do to circulating lactate concentrations?
- The fact that metabolic acidosis generated during exercise and other clinical conditions such as impaired oxidative phosphorylation mainly results from covalent modifications of glycolytic intermediates, such as phosphates, rather than from production of carboxylic acids, is an area of intense research. Another area of intense research concerns how protons actually function in oxidative phosphorylation. Despite Mitchell’s longstanding chemiosmotic theory in which protons are transported from the matrix into the intermembrane space and then move back through ATP synthase, controversies exist regarding the theory, including mechanisms of proton movement across the inner mitochondrial membrane. If you are interested, these controversies and related areas of research are summarized here – https://royalsocietypublishing.org/doi/10.1098/rsob.180221.
**NEW** – Finished with part I? Please see the update – Part II – Cellular Efflux of Protons and Lactate.
Selected Readings
- Forni LG, McKinnon W, Hilton PJ. Unmeasured anions in metabolic acidosis: unravelling the mystery. Crit Care. 2006;10(4):220. doi:10.1186/cc4954. PMID 16879718. https://ccforum.biomedcentral.com/articles/10.1186/cc4954
- Gevers W. Generation of protons by metabolic processes in heart cells. J Mol Cell Cardiol. 1977;9(11):867-874. doi:10.1016/s0022-2828(77)80008-4. PMID 592410
- Kraut JA, Madias NE. Lactic acidosis. N Engl J Med. 2014;371(24):2309-2319. doi:10.1056/NEJMra1309483. PMID 25494270.
- Krebs HA, Woods HF, Alberti KGMM: Hyperlactataemia and lactic acidosis. Essays Med Biochem 1975;1:81-103.
- Mizock BA. Lactic acidosis. Dis Mon. 1989;35(4):233-300. doi:10.1016/0011-5029(89)90021-7. PMID 2656163
- Morelli AM, Ravera S, Calzada D, Panfoli I. An update of the chemiosmotic theory as suggested by possible proton currents inside the coupling membrane. Open Biology. 2019;9(4):1-11. doi:10.1098/rsob.180221. PMID 30966998.
- Robergs RA, Ghiasvand F, Parker D. Biochemistry of exercise-induced metabolic acidosis. Am J Physiol Regul Integr Comp Physiol. 2004;287(3):R502-R516. doi:10.1152/ajpregu.00114.2004. PMID 15308499
- Robergs RA. Competitive cation binding computations of proton balance for reactions of the phosphagen and glycolytic energy systems within skeletal muscle. PLoS One. 2017;12(12):e0189822. Published 2017 Dec 21. doi:10.1371/journal.pone.0189822. PMID 29267370. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0189822
- Robergs RA, McNulty CR, Minett GM, Holland J, Trajano G. Lactate, not Lactic Acid, is Produced by Cellular Cytosolic Energy Catabolism. Physiology (Bethesda). 2018;33(1):10-12. doi:10.1152/physiol.00033.2017. PMID 29212886. https://journals.physiology.org/doi/full/10.1152/physiol.00033.2017?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
- Robergs RA. Invited review: Quantifying proton exchange from chemical reactions – Implications for the biochemistry of metabolic acidosis. Comp Biochem Physiol A Mol Integr Physiol. 2019;235:29-45. doi:10.1016/j.cbpa.2019.04.024. PMID 31071454.
- Vághy PL. Role of mitochondrial oxidative phosphorylation in the maintenance of intracellular pH. J Mol Cell Cardiol. 1979;11(10):933-940. doi:10.1016/0022-2828(79)90385-7. PMID 42805
- Zilva JF. The origin of the acidosis in hyperlactataemia. Ann Clin Biochem. 1978;15(1):40-43. doi:10.1177/00045632780150 PMID 24406. https://journals.sagepub.com/doi/pdf/10.1177/000456327801500111
This is fantastic. There are very few resources out there that compile all of this at once so learners are obliged to piecemeal it together with some inaccurate resources (which never works).
Question:
I’ve had a few patients with increased WOB, elevated lactate (~10-15 mM/L), and no acidosis due to paradoxical vocal chord motion which resolved with low dose bzd (a couple of them are “frequent flyers” with the same pattern each time). I’ve seen the same with increased work of breathing in asthmatics.
Have other people seen this phenomenon? I’m fairly certain I’m not seeing things 🙂
Is there any support for the idea that respiratory musculature has a greater predilection for production of lactate under high work load than other muscle?
It would seem that in cases with intact oxygen delivery, a case can be made for increased lactate production due to glycolysis while the proton load from ATP turnover can still be managed with oxidative phosphorylation. Do you think that could differentiate these cases from those with impaired DO2 as the main cause of both elevated lactate and acidosis?
Hi,
To answer this is to switch topics (assuming no hypoxemia) to when electron transport and oxidative phosphorylation are not systemically impaired. This would be analogous to intense exercise, and I’ll see if Rob will chime in in that regard. But during intense muscular work, ATP consumption exceeds that which can be produced by oxidative phosphorylation, while other organs and tissues are doing fine. I recall some particularly misleading papers in the critical care literature in the late 1970s and early 1980s on lactate production by diaphragmatic and intercostal muscles in COPD (COLD) and asthmatic patients. To complicate your scenario further, elevated catecholamines during times of intense stress can contribute to elevated lactate levels and metabolic acidosis through beta adrenergic receptor agonism. The exact reasons for this remain unclear, but we sure see it with epinephrine infusions, and I could go into the theories regarding Na/K-ATPase in skeletal muscle and sources of ATP utilization, etc, but that would really be getting off the topic. I’ll ask Rob if he might simply comment on hyperlactatemia during intense muscular work apart from medical illnesses. I know lactate levels during intense exercise certainly can exceed values you have described. I don’t want to speak too much about hyperventilation since that was a question for fellows!
Steve
I know that most patients I see with increased WOB in the ED, especially those with “wheezing” secondary to vocal cord dysfunction, invariably get albuterol thrown at them either prehospital or in the ED. In fact, I think lactates being drawn while someone puffed on an albuterol neb accounted for like half of the positive lactates at my former ED. Any chance that could be contributing in the cases you’ve encountered?
One other thought… Is part of the education problem here that we don’t really have a term for elevated lactate other than “lactic acidosis”? People don’t seem to use the word “lactatemia” very much. Should be we be referring to a “lactosis” and an “acidosis” separately?
Like most hospitals, my academic medical center reports plasma lactate levels as “lactic acid” levels. Does yours? That’s sort of misleading right off the start, though it certainly doesn’t affect patient care. And some academic references actually have it correct. In fact, take a look at Joel Topf’s “Nephrology Secrets” (4th edition) chapter on metabolic acidosis (which I just read last night for the first time). They also point out that protons come from ATP and that glucose to lactate is not proton-generating, as do references cited here. Rob has spent many years addressing the very issue you raise. Perhaps he will have something to say. But traditional teaching in physiology in medical school and non-medical school is to blame lactic acid, and this is reinforced by continued misleading statements found in standard biochemistry resources and countless other chapters and papers. I doubt this issue will be resolved in my lifetime. Perhaps Howard’s request for a post on this topic will have been a tiny step forward when historians look back on us 50 or 100 years from now.
Really cool explanation of the underlying biochemistry here! However, as someone who uses Stewards method in day-to-day practice, I have some questions.
You clearly explain how, from an electroneutrality point of view, lactate can be formed by reducing charged phosphate groups (by ATP formation). I’m not exactly sure to what degree all of these are strong anions but I’ll trust you that the math works out (it should, given the mathematical identity of Stewart & classical approach).
However, all of this holds up within the cell. Diagnosis of acid-base disorders is typically based on extracellular pH (as measured in plasma). This stoichiometry doesn’t hold up in the EC environment I think? In particular, phosphate is much less abundant there than lactate can potentially be. In that case, isn’t there still introduction of a strong anion in the extracellular space, resulting in acidosis? Or am I missing something?
Hi Maarten,
You are exactly correct that glycolysis and oxidative phosphorylation take place in the cell, while we are measuring circulating pH in blood and lactate in plasma. The formulae provided in figure 29 reflect what is going on in cytoplasm based on intracellular concentrations of substrates and products, competition among various cations for binding to negatively charged substrates using intracellular cation concentrations, adjustments for partial ionization at pH 7, etc. And you are correct that what we see in plasma reflects movement of protons and other ions, including lactate, in and out of cells with involvement of cotransporters, antiporters, ion channels, changes in pH, pulmonary CO2 elimination, expansion and contraction of extracellular volume, urinary excretion of lactate at high levels, bicarbonate buffers, etc. And as we know, intracellular pH can be different in cells of different organs and different than in blood.
The point of the post was to demonstrate that the –synthesis– of lactate by glycolysis does not result in significant changes in proton concentrations in cells that can be attributed to a significant dissociation of protons from lactate. This is in contrast to statements in various texts and countless papers that we form lactic acid from glucose and this lactic acid accumulates in the body to produce metabolic acidosis. This clearly is not true.
If a person comes up with additional pathways for the transport of lactate, protons, other ions, in and out of cells that may affect blood pH, those would not be changes due to lactate synthesis, which was meant to be the topic of this post. Certainly lactate contributes to an anion gap in plasma! But in balance, the protons that have entered plasma from the cell and caused a base deficit did not result from the synthesis of lactate. That is the message I am attempting to convey. None of us can even begin to imagine the complexities of all the simultaneous biochemical and structural cellular events that occur, even in a microsecond.
I’m going to ask Rob to comment on his thoughts regarding Stewart’s (I think that is who you were referring to) methodology and lactate. Take a look at reference #10 in the post – it includes a discussion of the Stewart approach. My very best regards.
Thank you for your answer!I did mistype Stewart’s name there. Certainly I’m not trying to argue that lactate formation is itself acidifying, just wondering whether it’s transport to the plasma might be – I’m not entirely ready to get rid of the term lactic acidosis (but I guess that is indeed a different topic).
Will you be providing answers to your questions at some point? These posts are always highly educational for those crit care nerds among us, who have only a general knowledge of toxicology.
Many thanks,
Maarten
I don’t know that the next post will be on transmembrane movement of lactate out of cells. I cautioned Howard that I was not sure anyone would actually read a post on the simplified biochemistry of lactate formation and impaired oxidative phosphorylation, but he thought otherwise, and was correct. Still, not sure how many would read about transmembrane transportation of lactate. I think I’ll return to more traditional toxicology, but may come back to this one day. Everything gets tied together in the end. I’ve never answered any of the questions I’ve brought up for fellows to discuss, so think I will keep with the tradition, at least at present. They will learn much more by arriving at the answers themselves. Thanks so much for commenting, it really is appreciated to learn that someone took the time to read the post.
Steve
Steven
think we need to talk. The sections on proton accounting from lactate production are excellent! However the conclusions in this post go way beyond that topic. You make a host of statements re: acid base in the plasma most of which are completely contradicted by all schools of acid base physiology (H/H and Stewarts included).
Everyone, Howard was correct, this has been a topic of much more interest than I ever anticipated, and I have been receiving emails asking about movement of lactate into and out of blood. Maarten and Scott, in comments below, raise valid points. In attempting to keep this simplified and not longer than it already is, transmembrane proton and lactate fluxes have not been addressed (and we know less about them). Acidosis in cells is not necessarily the same as acidemia. I am swamped at present but will either add either an addendum or long comment addressing this important aspect. Thanks!
Steve
Thanks for the interesting article. One factor (that has been raised previously in commentary of Dr Robergs’ work) is the reliance on the Henderson-Hasselbach model to look at pH changes. In Stewart’s model, lactate is an anion that will cause a rise in proton concentration from water dissociation. Is there a way to assimilate the concepts in your article with the Stewart model?
just look at fig 17 were we have electroneutrality
Very helpful! I have an additional question. Internists like to say that lactate is metabolized to bicarbonate by the liver. Looking at gluconeogenesis and the citric acid cycle, protons are formed in both. CO2 is certainly formed eventually but yields equal equivalents of bicarb and protons. Therefore, no net bicarb (unless the ETC consumes more protons than are formed) production can occur. What am I missing? Why is lactate (and citrate) called a
Bicarbonate precursor?
Hi. Sorry for the tardy reply but have been swamped, as have been all of us, I’m sure. Be sure to read the second section of this post. An answer to your question would require a very long response, but I will attempt to be brief. Just looking at proton balance, lactate to pyruvate to oxaloacetate and so on through gluconeogenesis does consume a proton (generate a HCO3-) if one counts the protons needed for ATP synthesis to fuel gluconeogenesis. And lactate going to pyruvate and then into the TCA cycle has been stated to consume protons or be relatively neutral – you generate one proton for each L to P, but consume protons in oxidative phosphorylation fueled by P. But these would not be immediate effects. When hypertonic Na lactate is given IV push, we must consider, for example, 1) the volume into which Na+ distributes; 2) the volume into which lactate distributes; 3) their rates of distribution; 4) whether tissues are normally perfused and taking up lactate, such as at rest, or whether shock/intoxication is causing lactate production; 5) if tissues are taking up lactate (including RBCs), then lactate moves intracellularly faster than Na+ leaves blood, changing the strong ion difference (I put that in there for the Stewart fans); 6) and if tissues are producing lactate and cannot take up what has been given IV immediately (other than RBCs), then lactate may not leave blood as rapidly and, theoretically, may not produce much of an immediate effect on pH; and 7) changes in blood volume from pulling water from cells and interstitium in response to the hypertonic load. And much more. For example when administered lactate is taken up by tissues, it can take a proton with it, can be exchanged for another strong anion like BOHB, can be exchanged for a chloride, etc., etc. The end result of all of these is that when hypertonic Na Lactate is compared to hypertonic NaCl, the Na Lactate bolus is associated with an initial rise in pH by some combination of mechanisms if the patient is not producing large amounts of lactate from shock/poisoning. To throw another wrench into things, Levraut and colleagues published a study about 22 years ago demonstrating that rises in lactate in patients with sepsis was mainly from decreased clearance, not from increased production. I haven’t meant to avoid answering, but just wanted to demonstrate how complicated things really are.
Best explanation of glycolysis and biochemistry I think I’ve ever read. Thanks
Thanks so much. If you like biochemistry, you may enjoy this one as well:
https://sandbox.emcrit.org/toxhound/ff-microvesicular-steatosis/
This is the best attempt I found for the biochemistry explanation for acidosis, associated with the formation of lactate in various medical conditions (which has been mistakenly associated for generations with the formation of lactic acid, hence “lactic acidosis”). I was looking for such a comprehensive biochemical explanation about the proton balancing in various acidosis conditions in the “established” literature for a long time.
Have you published it in the scientific review literature? If not, please consider it!
Thanks for this important document!
Thanks for kind words. Please be sure to read part II, as well.