, 1893. Public Domain from wikimedia.org](https://i0.wp.com/toxandhound.com/wp-content/uploads/2020/08/hero-image.png?resize=750%2C402&ssl=1)
{kind=link}
Cellular Efflux of Protons and Lactate
Steven C Curry, MD
University of Arizona College of Medicine – Phoenix
Banner – University Medical Center Phoenix
Phoenix, AZ
@SteveCurryMD
Robert A Robergs, Ph.D.
School of Exercise and Nutrition Sciences, Faculty of Health
Queensland University of Technology
Brisbane, QLD, Australia
As noted in the comments in the post on the origins of protons in lactic acidosis, I have received email inquiries regarding efflux of protons and lactate from cells during impaired oxidative phosphorylation, as well as how this fits in with the Stewart approach to acid/base disorders. I certainly was mistaken to attempt to simplify and keep short the original post on such a complicated topic, given more than a few thousand views. And it is true that what occurs in the cell cannot, in and of itself, be directly extrapolated, for example, to an anion gap or base deficit in blood without taking other factors into consideration. Thus, this “brief” addendum, again trying not to make things too long and trying not to get off the topic of the original post, which is that we generate lactate and not lactic acid during times of impaired oxidative phosphorylation.
Everyone must keep in mind that lactate production varies among different organs/tissues, even during times of systemic impairment of oxidative phosphorylation, such as hypoxia. Skeletal muscle at rest is the largest individual consumer of oxygen for ATP production, given its relatively large mass in the body and high ATP consumption, much of it used to power Na+/K+-ATPase in the sarcolemma. ATP requirements really take off during exercise or exertion. Muscle is a major, but certainly not the only, source of lactate during systemic inhibition of electron transport.
First, a quick word for medical toxicology fellows regarding membrane transporters. Very simplistically, and excluding ion channels and aquaporins, and ATPases such as Na+/K+-ATPase or Ca2+-ATPase, we can divide most protein membrane transporters into two groups. ATP-binding cassette proteins (ABC proteins) possess nucleotide binding domains where ATP binds, undergoes hydrolysis, and powers the transmembrane movement of molecules. In medical toxicology, the most common ABC protein mentioned is probably p-glycoprotein coded by the ABCB1 gene. Other ABC proteins combine with ion channels to control transmembrane ion fluxes – for example the KATP channel that regulates membrane potential in vascular smooth muscle and beta islet cells, or the Cl– transporter involved with the pathogenesis of cystic fibrosis. The second large group of membrane transport proteins are named solute carrier proteins (SLC proteins). SLC proteins do not bind and hydrolyze ATP, but rely on membrane potential and concentration gradients to power transmembrane fluxes of molecules. For example, Na+-dependent uptake pumps for neurotransmitters and various cation and anion transport proteins that transport drugs into hepatocytes for metabolism (e.g., OATs, OCTs) are SLC proteins. In this addendum, we will be looking at various SLC proteins that I have numbered 1-5 (my own numbering system) along the right side of the generic cell in Figure 33, below. Picking up where we left off, how do the protons from ATP hydrolysis and lactate generated from glucose or glycogen exit cells to enter blood?
Figure 33 shows protons generated from ATP hydrolysis and lactate anions produced from glucose. First, let us consider how lactate exits the cell. Monocarboxylate transporters (MCTs) compose a family of SLC proteins, of which four members are capable of transporting lactate anions (and other substrates). These transporters most commonly operate as cotransporters, meaning that they transport lactate or another negatively charged carboxylate along with a proton to maintain electrical neutrality.
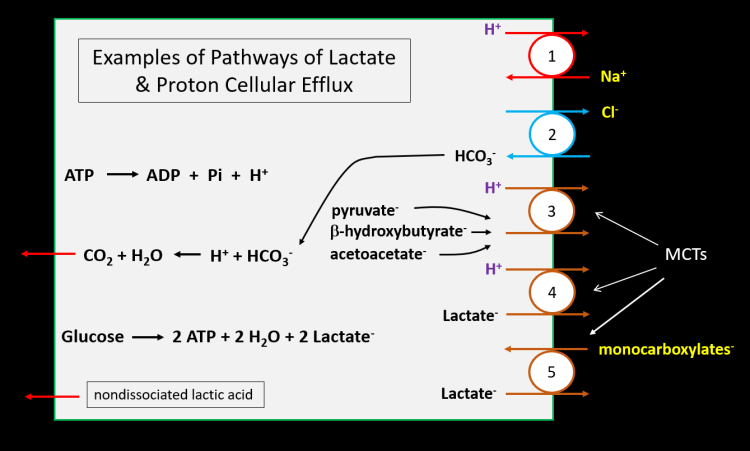
In Figure 33, SLC protein #4 represents an MCT cotransporting a proton with the lactate anion. Note that lactic acid is not transported. Of the MCTs that have been best modeled, studies show that a proton initially binds to its binding site, followed by lactate binding to a different site on the MCT. With conformational change, both the proton and lactate are moved to the other side of the membrane where lactate is released first, followed by release of the proton. A characteristic of MCTs and other SLC proteins is their reversibility. Some MCTs function normally to take lactate up into cells, such as for the Cori cycle or other processes. Even during severe lactic acidosis, erythrocytes take up lactate. However, these same transporters can reverse direction when intracellular proton and/or lactate concentrations rise high enough, as shown in Figure 33. In addition to lactate anions being transported with a proton, a portion of the small amount of intracellular nondissociated lactic acid may diffuse across the lipid bilayer of the plasma membrane out of the cell.
If lactate efflux only occurred when coupled to proton efflux, or as diffusion of nondissociated lactic acid, then stoichiometrically equal numbers of protons and lactate would enter blood (after traversing endothelium via various SLC proteins, including MCTs). We then would expect to see, at least initially, millimolar concentrations of circulating lactate matching those of base deficits and anion gaps if lactate and protons distributed into the same body fluid volume (ignoring lactate clearance and redox potential). Unfortunately, this is not the case. Both protons and lactate have other means of cellular egress, and protons distribute into a larger fluid volume than lactate anions. For example, at least one of the MCTs (#5 in Figure 33) that is expressed in most all cells can exchange lactate for another monocarboxylate anion, including acetoacetate, β-hydroxybutyrate, amino acid derivatives, fatty acids, and other compounds. When this occurs, lactate efflux takes place without efflux of a proton. Experiments also suggest that in some tissue lactate may be exchanged for Cl– or HCO3– by a non-MCT SLC protein. But most important are the other means of proton efflux. SLC #1 in Figure 33 shows a Na+/H+-exchanger that allows egress of protons without lactate. And the MCT illustrated as SLC #3 demonstrates that the same MCT that can transport lactate can, instead, cotransport protons along with various other monocarboxylates, with examples shown in the figure. Finally, SLC #2 in the figure shows that Cl– efflux is coupled to HCO3– influx. The imported HCO3– can combine with protons to produce CO2, which diffuses from the cell.
The end result is that when proton and lactate effluxes are studied in, for example, skeletal muscle, cellular proton efflux can be twice as large as that of lactate ions. This non-stoichiometric efflux of protons and lactate is a contributor to the often poor relationship between circulating lactate concentrations and changes in base deficits (using Copenhagen modeling of acid/base balance) and anion gaps. Once in blood, SLC proteins exchange or cotransport various ions across membranes of cells in various organs (including kidneys), and between plasma and blood cells, further modifying plasma ion concentrations. It is not surprising, then, that anion gaps usually are not explained completely by circulating lactate concentrations. In fact, the first paper I provided in the references explores the identity of the variety of organic anions responsible for anion gaps present during times of acidemia, even when lactate levels are elevated. Many of these are multiply charged anionic intermediates of the Krebs cycle.
So what do we end up with during systemic impairment of oxidative phosphorylation from decreased electron transport?
- Glucose to lactate occurs without generation of protons.
- The increased proton production responsible for metabolic acidosis mainly results from hydrolysis of ATP without ability to consume protons in oxidative phosphorylation.
- Lactate from glucose metabolism is cotransported with a proton from ATP hydrolysis out of cells, but not as lactic acid. In fact, the proton and lactate are released separately from the MCT. In some instances, lactate may be exchanged for a Cl– or HCO3–.
- Protons resulting from ATP hydrolysis egress from cells through various additional routes to a greater degree than lactate, especially from muscle, the individual organ responsible for most lactate production.
- We don’t produce significant quantities of lactic acid, and the non-stoichiometric relationship between cellular proton and lactate efflux accompanied by various other membrane ion fluxes contributes to the often-observed poor correlation between circulating lactate concentrations and changes in anion gaps. And don’t forget other factors that affect lactate levels, such as redox potential and lactate clearance, and that protons distribute into a larger space than lactate.
- Intracellular pH in skeletal muscle falls below 6.5 during intense exercise or hypoxia. As pH moves toward the pKa of lactate (3.67 @ 37 C and ionic strength 0.1 M), intracellular lactate will accept protons and slightly buffer changes in pH. This is factually true and even occurs in blood to a much lesser extent, where pH is higher. Lactate is not a major buffer of pH during times of acidemia, but it’s buffering action is noted for being the opposite of what is classically attributed to it – release of protons to lower pH.
Turning briefly to which ionic events or mechanisms explain a drop in blood pH in response to various ion fluxes across cell membranes during impaired oxidative phosphorylation, it is generally agreed that we possess reasonably good methods to predict values for pH and to categorize acid/base disorders, such as the Stewart and the Henderson-Hasselbach (H-H) approaches. But there remains uncertainty regarding the exact mechanistic causes of cellular and systemic pH changes – i.e., it remains difficult to isolate a single cause. As an example, if we take pure water (no buffers, no other ions) and add some hydrochloric acid (HCl) or some lactic acid, the addition of protons will cause a drop in pH. There are no other ions such as HCO3– present to influence proton concentration (pH). And if we run the ATPase reaction (ATP—>ADP + Pi + H+) in an unbuffered solution, pH steadily falls during ATP hydrolysis as protons are added to the solution (Figure 34). It is difficult, then, to imagine how proton efflux from cells into blood would not increase proton concentration and lower pH, just as protons do when added to water, especially since plasma is mainly water.

But plasma is more complex, filled with various ions and buffers. With the H-H approach, pH is predicted from HCO3– and pCO2, and protons that have egressed from cells raise blood proton concentrations and lower plasma [HCO3–], reflected in a decrease in pH. Since the protons described in this post originated from ATP hydrolysis rather than from lactate, elevated lactate concentrations would be considered unrelated to generation of acidosis or acidemia for those who see proton efflux as the primary event.
A segment of clinicians following the Stewart methodology advocate a main role of ionic neutrality in governing changing concentrations of multiple anions and cations in body fluids. In this approach, cellular proton efflux would not be considered important for purposes of predicting pH (i.e., proton concentration is considered a dependent variable). Rather the rise in plasma lactate concentration would narrow the strong ion difference and cause depression of plasma [HCO3–] and pH in order to maintain electrical neutrality in plasma. Just as hyperchloremia without accompanying elevations in strong cations such as sodium is accompanied by a lower plasma bicarbonate concentration and pH, so would hyperlactatemia, unless offset with a strong cation. In this situation, plasma lactate would be a contributor to metabolic acidosis via ionic neutrality, at least to the extent of the Δ-strong ion difference (SID) unexplained by Cl–. And there is mechanistic support for this approach, as well. The demand for charge neutrality negates a statement that elevated lactate concentrations during metabolic acidosis have no role in producing acidosis/acidemia, if both SID and bicarbonate levels are depressed.
With these observations in mind, let’s simplistically look at some examples of transmembrane ion fluxes from both standpoints, as some of you have requested, and examine immediate effects.
- efflux of cellular lactate exchanged for another strong monocarboxylate (anion): no change in plasma [H+]; no change in SID; everyone agrees, no change in pH, yet plasma lactate levels rise.
- efflux of cellular lactate exchanged for Cl–: no change in plasma [H+]; no change in SID; everyone agrees, no change in pH, yet plasma lactate levels rise.
- efflux of cellular lactate along with a proton: decrease in blood [HCO3–] and pH. Choose your mechanism: proton concentration rose during/after combining with bicarbonate to form H2CO3– and CO2; or the rise in lactate and fall in the SID depressed [HCO3–], lowering pH. Everyone ends up at the same spot.
- efflux of cellular protons exchanged for Na+: blood [H+] rises to cause fall in bicarbonate and pH, or SID falls (from sodium influx), which forces depression of [HCO3–] and pH. Plasma lactate unchanged. Everyone ends up at the same spot.
- efflux of cellular protons with another carboxylate anion: blood [H+] rises to cause fall in bicarbonate and pH, or the rise in blood carboxylate anion concentration causes SID to fall, which lowers [HCO3–] and pH. Lactate levels unchanged. We end up at the same spot.
From a mechanistic standpoint, both cellular efflux of protons which react with HCO3– and movement of ions to reestablish electrical neutrality in plasma must take place simultaneously. My general opinion is that both the Stewart method and the traditional H-H approach along with measurement of lactate levels and correcting the anion gap for albumin concentration generally perform the same in the ICU with sick patients with regard to outcomes. The Stewart approach has been reported to not do as well in calculating pH (proton concentration) during impaired oxidative phosphorylation from intense exercise-generated lactate efflux at blood pH < 7.26 or so, but this is a small effect from a single paper that probably is not clinically significant. Rob and I agree completely that, mechanistically, blood pH results from a complex mixture of factors, including 1) the ionization and dissociation constant of water, 2) circulating bases, 3) blood pCO2, 4) and metabolic H+ release, which can be dramatic during systemic impairment of oxidative phosphorylation. Regardless of which approach you use (and the truth may be some combination of both) to predict pH and categorize acid base disorders, we all end up with 1) elevated plasma lactate concentrations from proton-neutral generation of lactate, not lactic acid; 2) metabolic acidosis and, depending on other factors, acidemia; 3) changes in anion gaps and base deficits that commonly do not match circulating lactate levels, and most importantly 4) a recognition that we need to restore oxidative phosphorylation to correct the problem.
Medical toxicology fellows – one more question for you. I mentioned that SLC proteins can reverse transport under various conditions. For example, in the post on plant sodium channel openers, reversal of the 3Na+/Ca2+-exchanger explained Ca2+ influx during peaks of action potentials and as a contributor to arrhythmias from early afterdepolarizations in aconite toxicity. What role does reversal of neurotransmitter uptake pumps, also SLC proteins, play in intoxication by methamphetamine and/or MAO inhibitors? And why would cocaine prevent neuronal norepinephrine release induced by amphetamine?
Selected Readings
- Bangsbo J, Graham T, Johansen L, Saltin B. Muscle lactate metabolism in recovery from intense exhaustive exercise: impact of light exercise. J Appl Physiol (1985). 1994;77(4):1890-1895. doi:10.1152/jappl.1994.77.4.1890. PMID: 7836214.
- Dimmer KS, Friedrich B, Lang F, Deitmer JW, Bröer S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem J. 2000;350 Pt 1(Pt 1):219-227. PMID: 10926847.
- Fliegel L. Structural and Functional Changes in the Na+/H+ Exchanger Isoform 1, Induced by Erk1/2 Phosphorylation. Int J Mol Sci. 2019;20(10):2378. Published 2019 May 14. doi:10.3390/ijms20102378. PMID: 31091671.
- Halestrap AP. The SLC16 gene family – structure, role and regulation in health and disease. Mol Aspects Med. 2013;34(2-3):337-349. doi:10.1016/j.mam.2012.05.003. PMID: 23506875.
- Halestrap AP. Monocarboxylic acid transport. Compr Physiol. 2013;3(4):1611-1643. doi:10.1002/cphy.c130008. PMID: 24265240.
- Juel C. Muscle pH regulation: role of training. Acta Physiol Scand. 1998;162(3):359-366. doi:10.1046/j.1365-201X.1998.0305f.x. PMID: 9578382.
- Vijay N, Morris ME. Role of monocarboxylate transporters in drug delivery to the brain. Curr Pharm Des. 2014;20(10):1487-1498. doi:10.2174/13816128113199990462. PMID: 7836214.
Thanks for all of your hard work on this topic. I would really enjoy a podcast episode on this topic if that’s a possibility for you.
The way you explained with graphs is really informative. Keep sharing.
such a Nice blog share so unique content.
Avis Thermomix TM5 de Vorwerk